編者注:傅若農教授生于1930年,1953年畢業于北京大學化學系,而后一直在北京理工大學(原北京工業學院)從事教學與科研工作。1958年,傅若農教授開始帶領學生初步進入吸附柱色譜和氣相色譜的探索;1966到1976年文化大革命的后期,傅若農教授在干校勞動的間隙,系統地閱讀并翻譯了兩本氣相色譜啟蒙書,從此進入其后半生一直從事的事業——色譜研究。傅若農教授是我國老一輩色譜研究專家,見證了我國氣相色譜研究的發展,為我國培養了眾多色譜研究人才。
第一講:傅若農講述氣相色譜技術發展歷史及趨勢
第二講:傅若農:從三家公司GC產品更迭看氣相技術發展
第三講:傅若農:從國產氣相產品看國內氣相發展脈絡及現狀
第四講:傅若農:氣相色譜固定液的前世今生
第五講:傅若農:氣-固色譜的魅力
第六講:傅若農:PLOT氣相色譜柱的誘惑力
第七講:傅若農:酒駕判官—頂空氣相色譜的前世今生
第八講:傅若農:一掃而光——吹掃捕集-氣相色譜的發展
第九講:傅若農:凌空一瞥洞察一切——神通廣大的固相微萃取(SPME)
第十講:傅若農:扭轉乾坤—神奇的反應頂空氣相色譜分析
前言
脂質是一類自然界存在的疏水或兩性、難溶于水而易溶于非極性溶劑的有機物小分子,存在于大多數生物體系中。脂質是細胞膜的骨架物質和第二能量來源,還參與細胞的許多重要功能,人類許多重大疾病都與脂質代謝紊亂有關,如糖尿病、肥胖病、癌癥、阿茲海默癥、以及一些傳染病等,
作為代謝組學的重要分支之一,脂質組學(Lipidomics)的研究對象是生物體的所有脂質分子,并以此為依據推測其它與脂質作用的生物分子的變化,進而揭示脂質在各種生命活動中的重要作用機制。脂質組學是總體研究和這些疾病有關的脂質化合物,找到昭示這些疾病的生物標記物。
2005年國際上把組織、細胞中的脂質分子分為8大類(J Lipid Res 2009,50(Supp);9-14),有明確結構的脂質化合物已經有38000個(BMC Bioinformatics 2014, 15(Suppl 7):S9),這8類脂質分子見表1。
表 1 8大類脂質分子
|
類別 |
縮寫 |
數據庫中的結構數量 |
|
脂肪酰類(Fatty acyls) |
FA |
2678 |
|
甘油脂類(glycerolipids ) |
GL |
3009 |
|
甘油磷酸脂類(glycerophospholipids) |
GP |
1970 |
|
鞘脂類(sphingolipids ) |
SP |
620 |
|
固醇脂類(sterol lipids ) |
ST |
1744 |
|
異戊烯醇脂類(prenol lipids () |
PR |
610 |
|
糖脂類(saccharolipids ) |
SL |
11 |
|
多聚乙烯類(polyketides ) |
PK |
132 |
在過去,由于技術限制人們難以分析數量巨大的脂質分析,因為多種脂質代謝產物的物理性質需要大批純化系統、分離的復雜技術操作。2003年韓賢林等繼基因組學、蛋白質組學等之后提出脂質組學(lipidomics)(Han X et a1.J Lipid Res,2003,44:1071),脂質組學的發展推動了新分析平臺的研發,特別是在質譜法領域,該方法已使這些操作合理化,并且已允許更多的脂質分子得到非常詳細的分析。
脂質存在于細胞、細胞器和細胞外的體液如血漿、膽汁、乳、腸液、尿液中。若要研究某一特定部位的脂質,首先要將這部分組織或細胞分離出來。由于脂質不溶于水,通常采用有機溶劑進行萃取。傳統的萃取劑是氯仿、甲醇和水的混合液。所需的樣品在這種混合液中提取所有脂質,向提取液中加入過量的水使之分成2個相,上面是甲醇和水,下面是氯仿。脂質就留在氯仿相,蒸發濃縮后,使之干燥就得到所需的脂質。這種脂質提取方法,能夠提出組織樣品中的總脂。這種方法降低了脂質的損失率,操作簡便,而且提取效果較好。對于只檢測總脂中的部分脂質,固相萃取(SPE)是一種較好的方法,利用固體吸附劑將液體樣品中的目標化合物吸附,與樣品的基體和干擾物分離,然后再用洗脫液洗脫或加熱解吸附,達到分離和富集目標化合物的目的。固相萃取技術設備要求低,操作簡單,能快速分離組分復雜及含量低的樣品。當然由于化學分析樣品前處理技術的發展,有許多其他可用的樣品前處理方法。
總體上對脂質組學的研究Chin Chye Teo等歸納為如下的工作流程,第一步就是對樣品的處理。
1、脂質組學研究的工作流程
根據Chin Chye Teo的綜述報告(Chin Chye Teo et al,TrAC,2015,65:1-18),脂質組學研究的工作流程如下表1.
表1 脂質組學研究的工作流程
|
從患者得到脂質組學研究的樣品 |
|
|
液體 |
固體 |
|
體液,淚水,血清,血漿,尿液 (低溫保存樣品) |
細胞,組織,器官 |
|
對上述樣品進行萃取方法 |
|
|
對極性化合物,單獨的有機化合物進行: 液-液萃取,固相萃取 |
對能源性物質進行:加壓液相萃取,微波輔助萃取,超聲輔助萃取 |
|
萃取得到的脂質化合物 |
|
|
使用色譜方法分離:氣相色譜,液相色譜,電泳 |
不使用色譜方法分離:直接進樣,成像 |
|
上述分離或未分離樣品進行質譜分析 |
|
|
質譜分析的接口 |
質量分析器 |
|
電子轟擊電離(EI),電噴霧電離(ESI),化學電離(CI),大氣壓(APCI)化學與電離,基質輔助激光解析電離(MALDI) |
四級桿飛行時間質譜(qTOF),三重四級桿質譜( qqq),軌道阱質譜(Orbitrap) |
|
質譜原始數據語預處理 (利用商品或自制軟件) |
|
|
分類和脂質鑒定(使用各種資源如LIPID maps,Lipid Bank,Lipid Blast) |
|
|
判定在疾病中的機制/在疾病演化中的作用 |
|
|
為進一步診斷找出生物標記物(預防),提供藥物治療的指導 |
|
2、脂質組學的樣品制備
本文只講脂質組學的樣品制備,Chin Chye Teo等總結了近年在脂質組學研究中使用的樣品處理方法,見表2.
表2 脂質組學研究中的樣品處理方法比較(Chin Chye Teo et al,TrAC,2015,65:1-18)
|
萃取方法 |
臨床樣品類型 (生物液體或固體) |
優點 |
缺點 |
原文文獻編號 |
|
單一有機溶劑萃取(SOSE) |
血清(生物液體)
皮膚(固體) |
容易完成 萃取時間短 成本低 低溫適于熱敏感化合物 無需外部能量 |
使用有毒有機溶劑 分析時難以擺脫使用有機溶劑 |
1.2
3 |
|
液-液萃取(LLE) |
眼淚(生物液體) 血清(生物液體) 血漿(生物液體) 尿液(生物液體) 滑液(生物液體) 動脈粥樣硬化血小板(生物液體) 皮膚(固體) 組織(固體) |
易于建立的方法 容易完成 設備便宜 萃取時間短 使用廉價溶劑(如甲醇,水) 低溫適于熱敏感化合物 無需外部能量 萃取時間短 |
使用大量有毒有機溶劑 常使用超過一種類型的溶劑 需要排除溶劑以免影響分析 |
2 4,9-13 5,14-22 8,23 7 24
25-27 28,29 |
|
固相萃取(SPE)
|
血清(生物液體) 血清(生物液體) 血漿(生物液體) 眼(固體) 皮膚(固體) |
容易完成 清除干擾基體 EPE的選擇 低溫適于熱敏感化合物 萃取時間短 |
SPE萃取小柱比較貴 需要洗掉有機溶劑以免影響分析 使用有毒有機溶劑 分析時難以擺脫使用有機溶劑
|
1,12 2 30 26 3,27 |
|
固相微萃取(SPME) |
肺(固體) 頭發(固體) |
容易完成 可與GC和GC xGC 聯用 對揮發性化合物可以進行頂空氣相色譜 有毒溶劑消耗量少 低溫適于熱敏感化合物 無需外部能量 萃取時間短 |
萃取頭比較貴 需要洗掉有機溶劑以免影響分析 分析時難以擺脫使用有機溶劑 |
31 32 |
|
超臨界流體萃取(SFE) |
血漿(生物液體) |
容易完成 萃取時間短 對非極性化合物萃取效率高 CO2可循環使用 溫度壓力可控 可加改性劑提高萃取液極性和效率 |
要精心操作 設備昂貴 |
33 |
|
微波輔助萃取(MAE)
|
血漿(生物液體) 皮膚(固體) |
容易完成 萃取時間短 萃取效率高 萃取溶劑消耗量少 溫度壓力可控 |
需要冷卻防止溶劑逃逸 購買設備費用高 |
34
35 |
|
超聲輔助萃取(UAE) |
血(生物液體) |
容易完成 萃取時間短 萃取溶劑消耗量少 溫度壓力可控 |
聽力會受損 要使用有毒有機溶劑 會吸入有害溶劑 需要外部能源 購買設備費用高 提高溫度會使化合物降解 |
36,37 |
3、脂質組學的溶劑萃取
液-液萃取是脂質組學研究中使用最為普遍的方法,這一方法是使用兩種互不混溶的有機溶劑——使用最多的是氯仿、甲醇和水——為了對關鍵脂質類得到最大的萃取效率,從磷脂類和糖脂類到脂肪酸,三酰基甘油類(TAGs)、二酰基甘油類(DAGs)。最初使用的是Folch 脂質萃取法(氯仿/甲醇/水為 8:4:3 v/v/v),之后有Bligh 和 Dyer脂質萃取法(氯仿/甲醇/水為 1:2:0.8 v/v/v)。
(1)Folch 脂質萃取法(Folch et al., J Biol Chem 1957, 226: 497)
把樣品組織用2:1氯仿/甲醇均一化,最后的溶劑體積是組織的20倍(20mL 溶劑里有1g樣品),分散均勻后于室溫下把混合物在軌道振蕩器上震動15-20min。均勻混合物經漏斗中折疊濾紙過濾,或進行離心處理,回收液相。
液相溶劑用0.2體積的水(20 mL液相使用4 mL水),最好使用0.9%的NaCl溶液洗滌,渦旋幾秒后在低速離心機(2000 rpm)上離心混合物,用虹吸方法棄去上層液相,用以分析神經節糖苷或小分子有機極性化合物,如需要(需移去標記分子),用1:1甲醇/水洗滌交界處的有機相兩次,無需混合全部制備物。
經離心分離后虹吸掉上面的液相,下面含有脂質的氯仿在旋轉蒸發器中真空蒸發,或用氮氣吹拂到2-3 mL體積。
(2)Bligh 和 Dyer脂質萃取法(Can J Biochem Physiol 37:911-917)
a. 每1 mL 樣品加入3.75mL 1:2(v/v) CHCl3:CH3OH 很好渦旋,如果要進行GC 分析,溶劑中要含有內標(如0.5μg谷甾醇)
b. 然后加入1.5mL CHCl3很好渦旋
c. 最后加入1.25mL蒸餾水很好渦旋
d. 在1000rpm離心機中室溫下離心5min,得到一個兩相分離(上層為水相,下層為有機相)的液體
e. 回收有機相:用一個巴斯德吸管(Pastuer pipette)通過上層水相,輕微施加正壓避免上層水相浸入吸管,吸管口到達離心管底部,吸取下層有機相溶液的90%到吸管中。
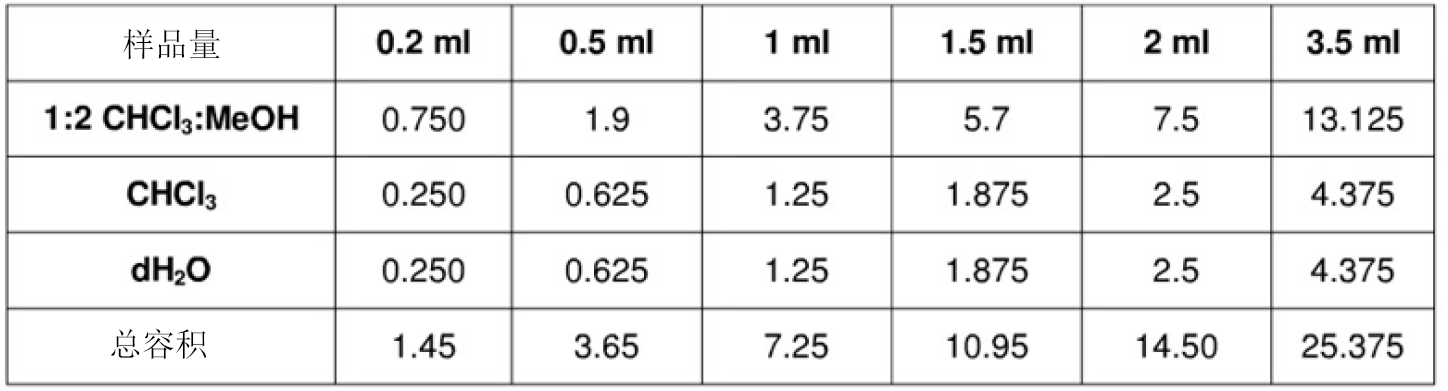
下表列出不同樣品容積需要加入的試劑量
如果你要得到干凈的底部的有機相溶液,就要用上層“真正”的上層液相洗滌有機相溶液,方法如下:
a 制備“真正”的上層液相:取一個大的玻璃管,或者幾個常規玻璃管,以水代替樣品胺上述方法進行萃取操作,把幾個管子中的上層水相合并在一起備用。
b 把上述第5步得到的底層溶液倒入一個玻璃管中,然后加入適量(樣品+蒸餾水的體積)“真正”的上層液相。比如你是1 mL樣品就加入2.25mL“真正”的上層液相。
c 好好地渦旋,離心,收集下層相。
Cui等的改進Bligh 和 Dyer脂質萃取法(Cui L,e al, PLoS Negl Trop Dis,2013,7:e2373):
900µL氯仿-甲醇(1:2)加入到100 µL樣品中,進行渦旋,在4°C下保溫,然后加入300µL氯仿和300µL雙重蒸餾水,以9000 rpm離心2 min,脂質物在離心管底部的有機相中,然后加入500 µL氯仿在4°C下進行渦旋20 min。從有機相中回收脂質物并與前次得到的脂質物合并,脂質萃取物經真空干燥后于−80°C下存放備用。
多少年來人們使用類似于上述方法進行脂質的萃取,例如:李國琛等在脂質組學研究中也采用Bligh 和 Oyer法萃取磷脂,并作適當改進.他們的方法是:
稱取100 mg魚肉樣品,加入400 p,L甲醇/氯仿(體積比2:1),渦旋混勻后,于一30℃放置過夜.取出后于4℃以10000 轉速離心5 min.將上清液轉出,在殘渣中加入200 mL甲醇/氯仿(體積比2:1)再次提取,將2次所得上清液合并.在上清液中先后加入100 mL氯仿及100mL水,離心后,將磷脂所在的氯仿相與水相分離.采用真空離心蒸發濃縮器干燥氯仿相(溫度不超過45℃,下同),將干燥后的樣品于一30℃保存備用.(高等學校化學學報,2010,31(2):269-273)
人們為了提高某些脂質種類的萃取效率,改變氯仿/甲醇/水的比例,并加入一些其他添加劑,如乙酸、鹽酸等,探索改進萃取各類脂質化合物的得率,如酸性磷脂和脂肪酸。(Jensen S K, Lipid Technol,2008, 20: 280–281)。
HCl-Bligh萃取法步驟:
為了更好地萃取生物樣品中的脂肪酸,使用加鹽酸的HCl-Bligh萃取法:取0.6 g均勻好的樣品裝入10-ml 帶蓋的培養試管中,加如1 ml 3M HCl,在80℃水浴上加熱1 h,之后加入1.50 ml甲醇和1.00 ml氯仿,以及17:0脂肪酸內標,把混合物搖震1 min,然后加入ELGA-純水系統制備的純水1.00 ml 和2.00 ml氯仿,把試管振蕩1 min,然后在3000 rpm離心機上進行離心處理5 min。把1 ml氯仿相進行甲基化,用氮氣把氯仿蒸發掉,加入0.8 ml NaOH/甲醇溶液,把試管充滿氮氣,密封在100 ℃下烘箱中15 min,冷卻后加入1 ml BF3溶液,密封在100 ℃下烘箱中45 min。在冷卻后加入2 ml辛烷和4 ml飽和NaCl溶液,把混合物進行渦旋,在3000 rpm離心機上進行離心處理10 min。用1μL 樣品進行氣相色譜分析。
根據Jensen的研究,認為此方法可以對脂肪酸的萃取率提高15%,對多不飽和脂肪酸的萃取率可提高30-50%。
由于氯仿的毒性大人們就用二氯甲烷來代替氯仿(J Agr Food Chem,2008,56:4297-4303),之后就有許多研究者效仿用以萃取臨床樣品,包括生物液體,如血清/血漿,尿液和固體樣品,如皮膚和動脈粥樣硬化血小板(表中文獻4,5,8,9,10,14-17,23-25,28).
近幾年也用甲基特丁基醚(MTBM )做萃取溶劑代替氯仿(Matyash et al. J Lipid Res. 2008,49 (5) :1137–1146.)。Matyash 認為MTBM進行萃取快速而且可以得到干凈的脂質,可以適合于自動進行鳥槍法得到脂質輪廓。因為MTBM的密度低,水相和有機相分開時,有機相在上層,這樣簡化了手機有機相的手續,減少了吸取的損失,不可萃取的基質小球處于離心管的底部,易于去除。嚴格的測試證明MTBM進行萃取對絕大多數脂質種類和“黃金標準”Folch 或 Bligh and Dyer萃取方法類似或更好。2013年中科院大連化學物理研究所許國旺和德國圖賓根大學醫學院的R Lehmannb使用MTBM進行萃取開創了一個從一小片肝臟或肌肉組織同時進行道謝組學和脂質組學的研究(J Chromatog A, 2013, 1298:9– 16)
人們的思路總是由簡單到復雜,又由復雜回歸到簡單,所以脂質組學中的萃取方法,近來也有多種溶劑向單一溶劑發展, Stübiger G (表中文獻1)就使用 Zhao Z等提出的單一溶劑萃取(SOSE)磷脂類脂質(J Lipid Res 2010;51:652)方法如下:
把500 mL甲醇加入到20 mL人血漿中,其中已經含有0.01% BHT(2,6-二叔丁基對甲酚)和0.5 mmol EDTA (用作抗氧化劑)和3mmol Pefablock(4-(2 aminoethyl) benzenesulfonylfluoride hydrochloride)用作磷脂酶的抑制劑,加入內標物,把樣品激烈震蕩1min,在冰浴中放置30 min,進行脂質的萃取,之后在10,000 rpm離心機上,離心5 min(4℃),最后把離心管上面的液體小心滴轉移到2 mL玻璃樣品瓶中,在零下70℃保存備用。
4、固相萃取(SPE)
SPE 是十分成熟的樣品預處理技術,使用裝有固定相的小柱子和各種流動相選擇性地保留與固定相有特定作用力的特殊種類分子。SPE的典型應用是和 SOSE 和 LLE相結合,作為一種附加的凈化步驟或從生物液體或固體住址樣品中富集某種特定種類的目標脂質(表中文獻1,3,12,26,27),市場有各種各樣的萃取小柱供選擇。供脂質萃取的SPE小柱有正相硅膠柱和反相柱(C8 和 C18),以及離子交換柱(氨丙基柱),硅膠柱和氨丙基柱多用于分離中性和極性脂質,利用改變洗脫溶劑以達到分離的目的。而C8 和 C18柱用于從水基樣品中分離卵磷脂(PC)、腦苷脂、神經節糖苷和脂肪酸。
針對不同的脂質使用不同的SPE,如 Stübiger(表2文獻1)在進行導致動脈粥樣硬化的磷脂的研究中,使用C18 凈化柱從血漿脂質萃取和富集體液氧化磷脂(OxPLs),其步驟如下:
把脂質萃取液倒入微量制備高效固相萃取柱(mHP-SPE)C18 spin-columns (PepClean, Pierce)中,小柱事先用500mL MeOH:0.2%甲酸(70:30 重量比)洗滌,然后用700 mL MeOH:0.2%甲酸(82:18 重量比)洗脫一次,再用800 mL MeOH:0.2%甲酸(92:2 重量比)洗脫一次,最后小柱用500 mL 2-丙醇再生,以便從小柱中徹底清除脂質(即中性脂質),凈化后的純度用薄層色譜檢查,得到的氧化脂質用LC-ESI-MS/MS進行分析。
而Ruben t’Kindt進行皮膚神經酰胺的脂質組學研究中,則使用氨丙基硅膠小柱對脂質萃取液進行凈化(表2文獻3),方法如下:
使用氨丙基硅膠小柱(100 mg, 3.0 mL)先用2 mL己烷洗滌,把已經干燥的脂質溶于300 μL 11:1 的己烷:異丙醇(v/v)中,用2 mL己烷/甲醇/氯仿(80/10/10 (v/v))洗脫神經酰胺,用氮氣吹掃干燥,溶于300 μL異丙醇/氯仿(50/50)(v/v)中,進行HPLC/MS分析。
5、固相微萃取(SPME)
Pawliszyn 研究組在1991年發明了SPME,1993年出現了SPME的商品化產品,使之成為廣泛使用的樣品前處理技術。這一方法是集萃取、濃縮、解吸、進樣于一體,它以固相萃取(SPE)為基礎,保留了SPE的全部優點,排除了需要柱填充物和使用有機溶劑進行解吸的缺點。SPME是以涂漬在石英玻璃纖維上的固定相(高分子涂層或吸著劑)作為吸收(吸附)介質,對目標分析物進行萃取和濃縮,并在氣相色譜進樣口中直接熱解吸(或用HPLC流動相沖洗到液相色譜柱中,甚至可以直接進行質譜分析),這一技術適合于揮發性和半揮發性有機物的樣品處理和分析。SPME有8大優點:1 操作簡單,2 功能多樣,3 設備低廉,4 萃取快捷,5 無需溶劑,6 可在線、活體取樣,7 可自動化, 8 可在分析系統直接脫附。SPME可以對環境中的污染物進行檢測,如:農藥殘留、酚類、多氯聯苯、多環芳烴、脂肪酸、胺類、醛類、苯系物、非離子表面活性劑以及有機金屬化合物、無機金屬離子等,也可以用有類似特點的領域,如食品、醫藥、臨床、法庭分析等方面。自然,在脂質組學中也會使用這一技術。
武漢大學曾昭睿研究組用自制的甲基丙烯酸丁酯/端羥基硅油萃取頭,萃取肺組織中的長鏈脂肪酸(表2文獻31)。F Pragst 利用SPME萃取頭發中的脂肪酸乙酯和葡萄糖苷酸乙酯來診斷過度酗酒(表2文獻32)。脂質中的脂肪酸都可以衍生化為酯類用SPME進行萃取。
SPME 的魅力在于它可以進行活體樣品中萃取分析物,用于代謝組學和脂質組學的研究,對這一課題SPME的發明人 Pawliszyn 近年進行了闡述(Angew Chem, 2013, 125:12346 –12348;Anal Chem, 2014, 86:12022−12029)。分析脂質代謝產物中游離脂肪酸的示意圖如下。
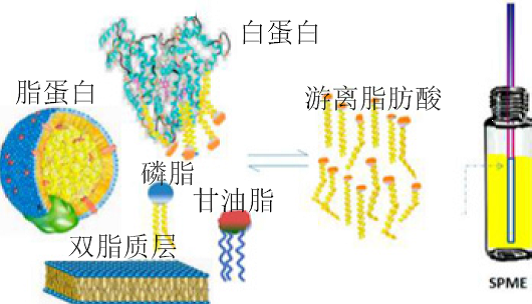
(Anal Chem, 2014, 86:12022−12029)
6、超臨界流體萃取(SFE)
超臨界流體具有特殊的理化特性,黏度為普通流體的1%~10%;擴散系數約為普通液體的10~100倍;密度比常壓氣體大100~1 000倍。因而超臨界流體既有液體溶解能力大的特點,又有氣體易于擴散和運動的特性,傳質速率大大高于液相過程。所以從萃取效率和對環境友好都受到歡迎。最常用的超臨界流體是超臨界二氧化碳(SF-CO2)它的臨界壓力和溫度低,只有7.4MPa和32℃。SF-CO2無毒易于從樣品中排除,其極性與戊烷近似,很適于萃取疏水性化合物,如脂質化合物(J Chromatogr A 2007,1163:2-24)。為了分離極性化合物往二氧化碳中加入改性劑,如甲醇。過去更多的工作時從植物類物質中萃取脂質,但是近來已經擴展到從動物組織中萃取脂質,例如浙江大學藥學院王龍虎利用江蘇省南通市華安超臨界萃取有限公司的 HA220-50-06 SFE裝置萃取鴕鳥脂肪中的脂肪酸:萃取裝置包括一個1 L 不銹鋼萃取釜,兩個1 L 分離器,一個注射泵,和一個冷凝裝置。用壓力調節器調節壓力,用可調節溫度的水浴控制溫度,通過調節泵的頻率來控制二氧化碳的流速。從液態二氧化碳鋼瓶把二氧化碳送到萃取器中,并達到超臨界狀態,在分離器中調節壓力和溫度可把萃取出來的組分里出來。試驗中取250 g鴕鳥脂肪組織用二氧化碳萃取5h,壓力15–30 MPa,溫度40–50℃,二氧化碳流速為15–35 L/h,用以考察萃取效果。(Eur. J. Lipid Sci. Technol. 2011, 113, 775–779)。
但是SFE更重要的是萃取人干血漿斑點中的脂質分子,Uchikata等(表2文獻33)比較了用SFE和液液萃取(Bligh 和 Dyer方法)磷脂的效果,證明SFE要比液液萃取方法對磷脂具有更好的選擇性,包括磷脂酰膽堿(PC)、溶血性磷脂酰膽堿(lysoPC)、磷脂酰乙醇胺(PE)和神經鞘磷脂(SM)。國內在1995年就有類似研究(薄層掃描法測定蛋黃磷脂中PC、SM和LPC的含量——路萍 賴炳森,藥物分析雜志,1995,(13):231-232),他們也是用SFE萃取之后進行薄層色譜分離。
7、微波輔助萃取(MAE)
微波輔助萃取(MAE)是利用微波能強化溶劑萃取效率,即利用微波加熱來加速溶劑對固體樣品中目標萃取物的萃取過程。MAE 可以快速高效地把樣品及溶劑中的偶極分子在高頻微波能的作用下,產生偶極渦流,離子傳導和高頻率摩擦,從而在短時間內產生大量的熱量。偶極分子旋轉導致的弱氫鍵破裂、離子遷移等加速了溶劑分子對樣品基體的滲透,待分析成分很快溶劑化,使微波萃取時間顯著縮短。
微波加熱具有選擇性微波對介電性質不同的物料呈現出選擇性的加熱特點,介電常數及介質損耗小的物料,對微波的入射可以說是“透明”的。溶質和溶劑的極性越大,對微波能的吸收越大,升溫越快,促進了萃取速度。而對于不吸收微波的非極性溶劑,微波幾乎不起加熱作用。所以,在選擇萃取劑時一定要考慮到溶劑的極性,以達到最佳效果。
MAE具有生物效應(非熱效應) ,由于大多數生物體內含有極性水分子,在微波場的作用下引起強烈的極性震蕩,從而導致細胞分子間氫鍵松弛,細胞膜結構電擊穿破裂,加速了溶劑分子對基體的滲透和待提取成分的溶劑化。因此,利用MAE從生物基體萃取待分析的成分時,能提高萃取效率。(李核等,分析化學,2003,31(109):126l~1268)
例如:萬益群,吳世芳利用MAE萃取何首烏中的磷脂(分析測試學報,2008,27(7):782—784),方法如下:確稱取約1.0 g何首烏樣品于溶樣杯中,加入20 mL萃取溶劑(氯仿與甲醇體積 比為1:2),把溶樣杯放入罐體中,組裝好罐體后放入微波制樣系統中,插入溫度探針。設置萃取壓力為安全壓力(1.5 MPa),萃取時間15 min,溫度為45℃。微波萃取完畢后,將樣品過濾。濾液用體積為濾液總體積l/4的8 g/L氯化鈉溶液萃取2次,收集有機相。將有機相旋轉濃縮至近干,用甲醇定容至10 mL。取樣品溶液3 mL用甲醇稀釋至10 mL,過0.45μm微孔濾膜,待測。
7、超聲輔助萃取(UAE)
超聲波為頻率高于20kHz以上的聲波,是一種機械振動在介質中的傳播過程,在傳播過程中,超聲波與介質的相互作用,可以使超聲波的相位和幅度等發生變化;功率超聲波則會使介質的狀態、組成、結構和功能等發生變化,超聲萃取中的應用可分為兩類:一類是頻率高,能量低(一般小于1W/cm2)的檢測超聲波,其頻率多以MHz為單位;另一類是頻率低,能量高(通常為10—100 W/cmz)的功率超聲波,其頻率則以kHz為單位。UAE是一種重復性好、萃取質量高的方法,它不像MAE,不會讓萃取系統的溫度升高,不利于熱穩定差的代謝物萃取。UAE還可以和液液萃取配合改進生物樣品中脂質的萃取效率。例如上海交通大學藥學院的劉玉敏等(Anal Bioanal Chem,2011, 400:1405–1417)成功地開發了UAE 和 LLE結合萃取人血清樣品中的代謝產物,從而比單獨使用液液萃取脂肪酸提高5–60%。Pizarro等使用類似的方法以MTBE作溶劑輔以UAE萃取人血中的脂質,比單純使用MTBE的液液萃取可以多檢出30%的脂質種類,MTBE-UAE萃取方法具有更好的重復性,相對標準偏差降低6%,脂質成分的回收率提高7成(表2文獻36)。除去萃取生物液體外,UAE-LLE也用于萃取樣品中的脂肪酸,例如哈爾賓醫科大學的李穎等研究了用UAE-LLE萃取鼠的肝臟組織,考察了超聲波功率、萃取溶劑、萃取容積、萃取時間等,結果表明萃取時間比Folch萃取法萃取脂肪酸從12 h 縮短到 20 min,回收率在87–120%之間。(J Chromatogr Sci, 2013;51:376–382)
8、其他可用的萃取方法
在化學分析樣品處理中還有兩種重要的樣品前處理方法,即加速溶劑萃取(ASE)和基質固相分散萃取(MSPD),可以用于脂質組學研究的樣品前處理。
加速溶劑萃取(Accelrated Solvent Extraction, ASE),這一方法是一種在提高溫度和壓力的條件下,用有機溶劑萃取的自動化方法。與其他液體萃取方法相比,其突出的優點是有機溶劑用量少、快速、回收率高。(牟世芬等,現代分析儀器,2001,(3):18-20)。 Spiric A等使用ASE萃取鯉魚肉中的脂肪酸譜和膽固醇含量,并與改進的索氏萃取法進行比較,表明ASE萃取方法是可用的。(Anal Chim Acta,2010, 672:66–71)。Jansen B等利用ASE從土壤中萃取脂質生物標記物,萃取效果和其他萃取方法一樣(Appl Geochem ,2006, 21:1006–1015)。Balasubramanian R K等用ASE和其他方法進行了從海水微海藻細胞中萃取脂質的研究,表明ASE是一種可以使用的方法(Chem Engineering J,2013, 215–216:929–936)。
MSPD方法是1989年首次提出是用來處理動物組織樣品的方法,樣品與涂漬有C18等的各種聚合物載體的固相萃取材料一起研磨,得到半干狀態的混合物并將其作為填料裝柱,然后用不同的的溶液洗脫柱子,將各種待測物洗脫下來。其依據是采用脂溶性材料(C18)破壞細胞膜并將組織分散,C18充當分散劑。在硅膠固相萃取材料表面鍵合有機相,與傳統方法使用砂子做吸附劑類似,在樣品與固體材料攪拌的過程中,利用剪切力作用將組織分散。鍵合的有機相就像溶劑或洗滌劑一樣,將樣品組分溶解和分散在支持物表面。這大大增加了萃取樣品的表面積,樣品按各自極性分布在有機相中,如非極性組分分散在非極性有機相中,極性小分子與硅膠上的硅烷醇結合,大的弱極性分子則分散在多相物質表面。(烏日娜等,食品科學,2006,26(6):266-268)。香港城市大學的Qing Shen等利用二氧化鈦納米顆粒作萃取劑,以基質固相分散萃取方法進行橄欖果的脂質組學研究,研究證明這一方法可以把磷脂從非磷脂中完全選擇性地分離出來。(Food Research Int,2013, 54:2054–2061)。
表2中的文獻
|
1 |
Stubiger G, et al, Atherosclerosis, 2012,224:177–186. |
|
2 |
Zhao Z, et al, J Lipid Res, 2010, 51:652–659 |
|
3 |
t’Kindt R, et al, Anal Chem, 2012,84:403–411 |
|
4 |
Cui L, et al, PLoS Negl Trop Dis,2013,7:e2373 |
|
5 |
Sandra K,et al, J Chromatogr A,2010,1217:4087–4099. |
|
6 |
Lam S M, et al, J Lipid Res, 2014,55: 289–298 |
|
7 |
Giera M, et al, Biochim Biophys Acta, 2012, 1821:415–424 |
|
8 |
Min H K, Anal Bioanal Chem, 2011, 399:823–830. |
|
9 |
Heilbronn L K, et al, Obesity,2013, 21:E649–E659 |
|
10 |
Hilvo M, et al, Int J Cancer 134 (2014) 1725–1733 |
|
11 |
Montoliu I, et al, Aging (Albany NY),2014,6:9–25 |
|
12 |
Chen Y , et al, Clin. Chim. Acta, 2013,428: 20–25. |
|
13 |
Zivkovic A M, et al, Metabolomics,2009,5:507–516 |
|
14 |
Chen F,et al, Biomarkers, 2011, 16:321–333 |
|
15 |
M. Ollero, et al, J. Lipid Res, 2011, 52:1011–1022 |
|
16 |
Shah V, Rapid Commun. Mass Spectrom, 2013, 27:2195–2200 |
|
17 |
Lankinen M, et al, PLoS ONE, 2009,4:e5258. |
|
18 |
J. Graessler, et al, PLoS ONE,2009, 4:e6261 |
|
19 |
Lofgren L et al,, J Lipid Res, 2012,53:1690–1700 |
|
20 |
Gurdeniz G, et al, PLoS ONE, 2013,8:e69589. |
|
21 |
Zhou X, et al, PLoS ONE, 2012, 7:e48889. |
|
22 |
Bui H H, et al, Anal Biochem, 2012,423:187–194. |
|
23 |
Kim H, et al, Analyst, 2008, 133:1656–1663. |
|
24 |
Stegemann C, et al, Circ Cardiovasc Genet, 2011,4:232–242. |
|
25 |
van Smeden J, et al, J Lipid Res, 2011,52:1211–1221. |
|
26 |
Acar N, et al, PLoS ONE,2012, 7:e35102. |
|
27 |
Shin J H, et al, Anal Bioanal Chem,2014,406:1917–1932 |
|
28 |
Cheng H, et al, J Neurochem, 2013,127:733–738. |
|
29 |
Pietilainen K H,et al, PLoS Biol,2011, 9:e1000623. |
|
30 |
Cha D, et al, J Chromatogr A,2009,1216:1450–1457. |
|
31 |
Cha D, et al, Anal Chim Acta,2006, 572: 47–54. |
|
32 |
Pragst F, et al, Forensic Sci Int,2010, 196: 101–110 |
|
33 |
Uchik T,et al, J. Chromatogr A, 2012,1250:69–75. |
|
34 |
de Morais D R, et al, Rev Bras Hematol Hemoter,2010,32:439–443. |
|
35 |
Gonzalez-Illan F,et al,J Anal Toxicol,2011,35:232–237. |
|
36 |
Pizarro C, et al, Anal Chem,2013,8:12085–12092. |
|
37 |
Pang L Q, et al, J Chromatogr B,2008,869: 118–125 |