高效液相色譜技術(shù)最早應(yīng)用于藥物領(lǐng)域,包括效能/純度/性能分析、藥物(代謝)動力學(xué)/生物分析測試、純化、高通量篩選(HTS)、過程控制(IPC)監(jiān)測和質(zhì)量控制(QC)測試。制藥行業(yè)是HPLC的主要應(yīng)用市場,同時也是HPLC向更高通量和更優(yōu)性能發(fā)展的主要驅(qū)動力。本文簡要回顧了過去十年影響藥物分析發(fā)展的HPLC的重大進展。討論的主題如下,部分項目體現(xiàn)在一個典型的時間軸上,如圖1所示。
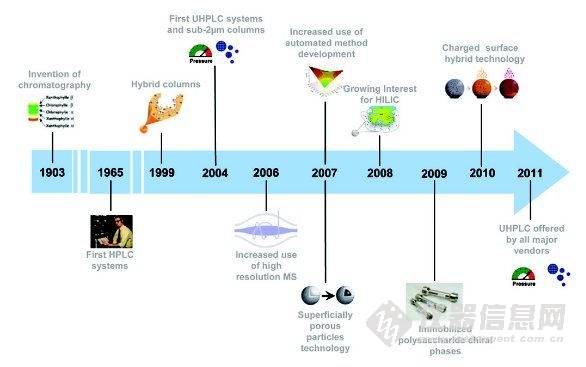
圖1 過去十年中影響藥物分析發(fā)展的HPLC技術(shù)進展(部分項目)
本文的目的是為繁忙的制藥科學(xué)家提供一個簡潔又全面的HPLC的重要的技術(shù)進展,每個主題都有實際使用過程中的便利和應(yīng)用的簡短描述,從用戶的角度的評論,以及主要參考文獻(略)的支撐。
超高壓液相色譜(UHPLC)
UHPLC的革命性創(chuàng)新始于1997年James Jorgenson教授在概念驗證方面的研究,緊接著第一個商業(yè)化系統(tǒng)于2004年推出。今天,從HPLC到UHPLC的轉(zhuǎn)化大部分都是由主要制造商完成,這些制造商目前都可以供應(yīng)某種類型的UHPLC產(chǎn)品。UHPLC系統(tǒng)、柱子以及應(yīng)用的詳細綜述在其他地方都可以看到。UHPLC在藥物分析領(lǐng)域的基本原理、優(yōu)勢、潛在的問題和最佳應(yīng)用都有充分的文件記錄。
表1概述了UHPLC的突出特點和優(yōu)勢。注意,較高的系統(tǒng)壓力允許使用更小的微粒填充的柱子(如亞2μm)得到更快的分析速度(圖2)或復(fù)雜樣品更出色的分離(圖3)。由于使用改良的進樣器,內(nèi)徑較小的管路系統(tǒng)(< 0.005英寸),更小的紫外檢測器流動池(0.5 - 2μL),所有的UHPLC都有比較低的系統(tǒng)擴散(4σ時,~10-20μL)。比較典型的應(yīng)用是內(nèi)徑為2.1–3.0mm的柱子,填充物為亞2μm或亞3μm顆粒。其他重要的系統(tǒng)特征包括更小的系統(tǒng)死體積(0.1-0.3 mL)和更快的檢測器響應(yīng)/數(shù)據(jù)采集速率(>40 pt/s),以實現(xiàn)高通量的應(yīng)用。
表1 UHPLC的突出特點和優(yōu)勢
|
UHPLC的系統(tǒng)特點 |
范圍和評論 |
|
最高壓力限制 |
15000到19000psi(1000到1250bar),流速2-5mL/min.兼容傳統(tǒng)的和亞-2微米顆粒的色譜柱。 |
|
低系統(tǒng)色散 |
根據(jù)儀器配置,儀器帶寬5-20μL(4σ),使用較小的連接管(<0.005"I.D)和小UV流通池(0.5-2μL),減小系統(tǒng)頻帶展寬。兼容ID低至2-3mm的色譜柱。 |
|
低的梯度保留體積 |
100-400μL(對于四元液相泵會更高),兼容高通量篩選(HTS)。小的保留(混合)體積可能影響UV檢測器噪聲。 |
|
其他 |
HTS快速的注射周期(-20s)和檢測響應(yīng),以及高的采集率(>40pt/t),兼容目前的HPLC方法需求(如流量范圍、柱溫箱尺寸、進樣環(huán)路) |
|
UHPLC的優(yōu)勢 |
評論 |
|
高通量 |
在保持相似分辨率的情況下,與傳統(tǒng)的HPLC方法相比通量提高3-10倍。如純度分析5min(UHPLC)/20min(HPLC)。 |
|
快速方法開發(fā) |
短色譜柱,快速分析是色譜柱和流動相快速篩選及方法優(yōu)化的理想選擇。 |
|
高分辨 |
相對于HPLC,分辨率提高3倍,比如峰容量(Pc)400-600(HPLC為200) |
|
溶劑節(jié)省 |
分析時間短,使用較小ID的色譜柱,相比HPLC溶劑節(jié)省5-15倍。 |
|
高靈敏度 |
質(zhì)量靈敏度增加3-10倍(樣品注入量減少)。長路徑UV流通池(50-60min)可以將濃度靈敏度提高6倍。 |
|
高精度 |
保留時間(2-3倍)和峰面積精密度(<1%RSD,進樣體積>1μL )顯著增加 |
|
可以與其他的方法聯(lián)用 |
UHPLC兼容高溫LC,2D-LC或者核殼色譜柱(單一或者組合聯(lián)用)。這些都是可選項,而不是“有它就不能有它”。 |
與傳統(tǒng)的HPLC相比,UHPLC具有分析速度快的優(yōu)勢,圖2闡述了一種藥物分析由HPLC方法轉(zhuǎn)移為UHPLC方法的譜圖對比。從HPLC到UHPLC,根據(jù)幾何尺寸按比例縮放色譜柱及操作參數(shù),可以在相同分辨率的情況下將分析時間減少10倍,這并不罕見。“保證好的分辨率的情況下進行更快的分析”這是大多數(shù)用戶考慮購買更昂貴的UHPLC設(shè)備的主要誘因。
UHPLC的另一個重要的好處是其對復(fù)雜樣品優(yōu)秀的分離能力。這方面常常被忽視,在文獻中也很少報道。在合理的時間跨度(~1 h)內(nèi),400至1000范圍內(nèi)的峰容量(PC)已經(jīng)通過使用UHPLC來證實。峰容量是分辨率為1.0時,色譜圖上可以分辨的色譜峰個數(shù);通常傳統(tǒng)的HPLC為200左右。有史以來第一次,UHPLC可以在單一維度,對復(fù)雜的藥品、天然材料、和其他困難樣品基質(zhì)提供更有效的分析。圖3a和3b通過兩個應(yīng)用 (植物提取物和胰蛋白酶的消化蛋白質(zhì))來說明Pc>300時,當(dāng)前UHPLC的高分辨分離能力。
UHPLC的其他優(yōu)勢包括溶劑節(jié)省(5-15倍),增強的質(zhì)量靈敏度(3-10倍),以及在保留時間(2–3倍)和峰面積(<0.1%RSD)方面的卓越性能。注意,有關(guān)UHPLC中UV檢測靈敏度增加的報告往往會產(chǎn)生誤導(dǎo),因為質(zhì)量敏感度(分析物注入量)主要與柱的空隙體積有關(guān)。通常,UHPLC不會增加濃度靈敏度(最理想的一種靈敏度),因為我們不能希望使用小的流通池來增加信噪比,除非延長流通池的路徑長度 (如60毫米)。泵混合而導(dǎo)致的粘性發(fā)熱、基線擾動以及方法轉(zhuǎn)移等潛在問題已經(jīng)有文獻描述,而且這些問題分別也是與特定的儀器相關(guān)聯(lián)的。一般來說,這些技術(shù)問題已經(jīng)研究得比較清楚了,而且通過選擇合適的系統(tǒng)配置(如混合器體積)能夠很容易地減輕。但是, 由于培訓(xùn)、與驗證數(shù)據(jù)系統(tǒng)的兼容性和其他方法轉(zhuǎn)移的問題,UHPLC在QC實驗室的應(yīng)用仍然比較浪費時間。
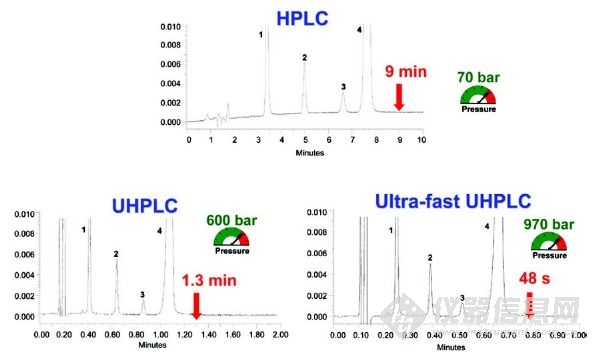
圖2 以商業(yè)制藥配方(Rapidocain) 質(zhì)量控制分析為例:從傳統(tǒng)的HPLC到UHPLC使用幾何比例縮放。峰識別:1:對羥基苯甲酸甲酯,2:2、6-二甲基苯胺,3:尼泊金丙酯,4:利多卡因。HPLC條件:色譜柱RP18 150 x4.6毫米,5μm, F=1ml/min, Vinj=20μL。UHPLC條件: 色譜柱RP18 x2.1 50毫米,1.7μm,F = 600μL /min,Vinj = 1.4μL。超快UHPLC條件:色譜柱RP18 x2.1 50毫米,1.7μm,F = 1000μL /min,Vinj =1.4μL[16]。
我們經(jīng)常聽到一些爭論,UHPLC也許并不需要,因為其他方法(高溫LC、核殼柱子或二維LC)可以更合算的提高速度或分辨率。這種推理并不是真實有根據(jù)的,因為與傳統(tǒng)的HPLC相比,UHPLC在使用過程會中可以與一個或多個以上的這些方法聯(lián)用獲得更出色的結(jié)果。這些方法都是可選項,而不是“有它就不能有它”。此外,“UHPLC”這個詞語可能在幾年內(nèi)最終消失,因為所有新的HPLC都是UHPLC了。
HPLC色譜柱和固定相的發(fā)展
HPLC色譜柱是色譜系統(tǒng)的核心。在堿性分析物分析方面,制藥業(yè)一直是HPLC色譜柱向高速、高分辨率、更好的峰形發(fā)展的主要驅(qū)動力。此外,QC實驗室已經(jīng)被要求改進色譜柱的批次重現(xiàn)性。從70年代到90年代,在填充材料的標準顆粒尺度(10-3μm)的逐級縮小方面已經(jīng)有穩(wěn)定的改進。80年代后期,高純度B型硅材料(低金屬含量)的引入是一個巨大進步,減少了硅醇的活性,并在批次間的一致性方面有重大改進。現(xiàn)在高純硅的使用是所有現(xiàn)代硅膠基質(zhì)色譜柱的標準。
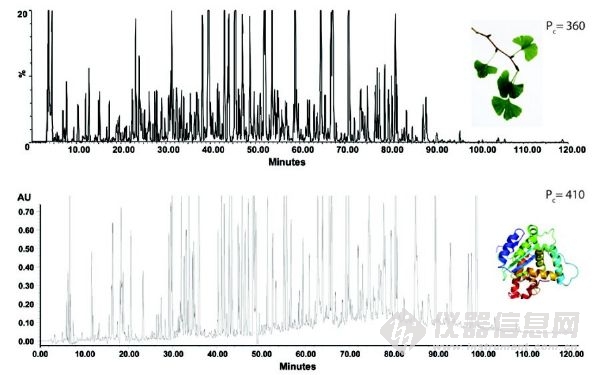
圖3 UHPLC 高分辨率分離(C18色譜柱,填充亞2μm顆粒)。(A)銀杏標準化提取物的色譜圖。梯度:5%到40%的乙腈,120分鐘,T=30°C, Lcol = 300mm,F(xiàn) = 0.2 mL/min,ΔPmax = 860 bar,峰容量可以達到360;(B)三種蛋白質(zhì)的結(jié)合胰蛋白酶消化酶的色譜圖。梯度:5%到60%的乙腈,120min ,T=30°C,Lcol = 40cm,F = 0.16 mL/min,ΔPmax = 940bar,峰容量可以達到410。
HPLC色譜柱技術(shù)的近期進展已經(jīng)被廣泛地綜述(已經(jīng)有很多文獻綜述并討論了HPLC的色譜柱技術(shù)的近期進展)。表2總結(jié)了重大行業(yè)使用率較高的,用于提高生產(chǎn)力、穩(wěn)定性、選擇性或保留時間(亞2μm,核殼粒子,混合物,新奇的鍵合反應(yīng),HILIC)或?qū)I(yè)應(yīng)用 (手性化合物或生物分子)的色譜柱。
表2 影響藥物分析的重要的HPLC色譜柱的研究進展
|
HPLC色譜柱重要研究進展 |
評論 |
|
亞2μm顆粒 |
全孔亞2μm顆粒產(chǎn)生較低的塔板高度和高的塔板效率。10多家供應(yīng)商提供超過80種可用的化學(xué)反應(yīng),包括離子交換、尺寸排阻等。 |
|
亞3μm核殼顆粒 |
表面多孔型亞-3μm顆粒在低柱壓條件下提供改善的質(zhì)量傳遞和較低的塔板高度。增加了在藥物分析中的使用案例。 |
|
混合硅顆粒 |
創(chuàng)新的顆粒提高色譜柱的化學(xué)穩(wěn)定性(擴大pH范圍、溫度和壓力性能,親硅醇基活性) |
|
新奇的鍵合化學(xué)反應(yīng) |
多官能團的硅烷化學(xué)、極性嵌入式鍵合相、五氟苯酚(PFP)、 苯基-己基、表面帶電雜化(CSH)相,提供“正交”分離和增強的選擇性(與傳統(tǒng)的C18鍵合相相比) |
|
親水作用色譜(HILIC) |
反相色譜的“正交”分離對強極性化合物和次生代謝物有用 |
|
用于手性分離的多糖固定相 |
多糖手性固定相使得通常的色譜柱具有多用性和抗造性 |
|
生物分子色譜柱 |
創(chuàng)新的核殼結(jié)構(gòu)和UHPLC大孔徑固定相可以有效改善蛋白質(zhì)和治療性生物分子(如mAbs)的分析。 |
亞2 μm顆粒
1956年,Van Deemter就給出了使用非常小的顆粒進行快速、高效分離的預(yù)測。在過去五年中,典型填料的粒徑在不斷的減小,二十一世紀初,集中在亞2μm的硅微顆粒。正如預(yù)測的那樣,這些粒子(例如,1.7μm)產(chǎn)生卓越的性能(~ 280000 plates/m或~ 4μm/plate)。然而,填充亞2μm粒子的色譜柱產(chǎn)生高的背壓,通常填充內(nèi)徑為2.1毫米,通過粘性發(fā)熱減少效率損失。對高壓力和低分散(減少附加柱的譜帶展寬)系統(tǒng)的要求導(dǎo)致了現(xiàn)代UHPLC系統(tǒng)的當(dāng)前特點。進一步降低粒徑至小于1.5μm可能會產(chǎn)生更高的速度和性能。然而,它也必須伴隨著系統(tǒng)壓力的大幅增加和毛細管色譜柱內(nèi)徑的減小。
核殼結(jié)構(gòu)顆粒
Kirkland第一次描述了熔融的核或者核殼粒子的概念,減少質(zhì)量轉(zhuǎn)移過程中的阻力。第一個核殼粒子具有如下特點:2.7μm表面多孔硅材料,無孔的核心(1.7μm),多孔的殼層(厚0.5μm)。這些亞3 μm的顆粒與亞2μm的完全多孔材料相比似乎具有相似的效率,但是可以產(chǎn)生更低的壓降。這種特殊的性能可能是由于殼體較短的擴散路徑,或者比較狹窄的填料分布。由于快速分離(HIS、IPC和清潔驗證)和在生物分子方面的應(yīng)用,核殼色譜柱迅速獲得廣泛的接受。越來越多的制造商可以提供各種鍵合相和不同尺寸的粒子(1.3,1.7,2.6,1.3和5μm)。我們完全相信,與多孔微細顆粒的色譜柱相比,這些色譜柱在所有應(yīng)用中具有很大的競爭力。
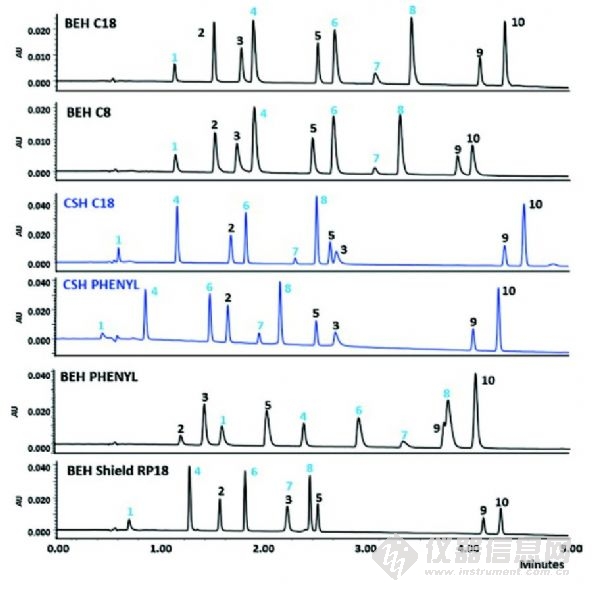
圖4 10種具有酸性、堿性和中性官能團的商品化藥物在BEH和CSH分析柱的分析比較,梯度洗脫,流動相含乙腈和0.1%的甲酸。峰1、4、6和8是基本藥物。從文獻中復(fù)印,已取得許可。
雜化
將有機基團引入到無機硅基體中形成雜交顆粒的理念在上世紀70年代首次由Unger提出,雖然第一根具有甲基基團的商業(yè)化的色譜柱在1999年才正式推出。這些混合物中的鍵合相被證明具有很好的pH穩(wěn)定性(具有新奇的鍵合化學(xué)反應(yīng)的pH范圍為1到12,而傳統(tǒng)的具有常規(guī)單功能鍵合硅顆粒的pH范圍為2-8),低的親硅羥基活性。2005年,引入第二代橋聯(lián)乙烯雜化 (BEH),并立即獲得了“主流”成功,特別是高pH值流動相以及UHPLC的應(yīng)用方面。
新奇的鍵合反應(yīng)
或許是因為批次之間重現(xiàn)性最好,傳統(tǒng)的單官能團的C18硅膠基質(zhì)鍵合相仍是主要的產(chǎn)品,但是新的鍵合化學(xué)反應(yīng)給困難的分離(極性嵌入式、苯基、己基苯基、含氰基的、戊烷氟苯基鍵合)帶來了更寬的pH值穩(wěn)定范圍(多官能的硅烷化學(xué)鍵或異丙基保護的硅烷)和增強的選擇性。最近的一個創(chuàng)新方法稱為表面帶電雜化(CSH)技術(shù),該項技術(shù)于2010年引入,由于在酸性,低離子強度流動相條件下,對高堿性分析物的峰形的改善,該項技術(shù)立即在藥物分析領(lǐng)域獲得了很好的接受度(如0.1%甲酸)。這項專有技術(shù)包括在固定相表面引入低水平的正電荷。這類似于早期在流動相中添加有機胺,如三乙胺,但由于離子抑制,已經(jīng)不被LC/MS接受。圖4說明了與現(xiàn)在的鍵合相相比,CSH顆粒在峰形方面表現(xiàn)的優(yōu)勢。注意,酸性化合物在CSH柱子上會有相對較高的保留,峰拖尾現(xiàn)象也會有所增加。
親水作用色譜(HILIC)
在反相LC(RPLC)條件下,若流動相中有機物含量比較低,就會導(dǎo)致相坍塌現(xiàn)象(鍵合相脫水),許多強極性化合物就無法獲得足夠的保留時間或者會存在問題。HILIC模式, 90年代由Alpert首次開發(fā),使用親水固定相(硅、二醇、氰基、氨基、兩性離子的),類似RPLC使用的水緩沖和乙腈流動相,在極性藥物分析、輔助藥物代謝、氨基酸、多肽、神經(jīng)遞質(zhì)、低聚糖、碳水化合物、核苷酸或核苷方面的分析中越來越受歡迎。HILIC實際的保留機制可以認為是分析物分子“分區(qū)”到附著在親水結(jié)合基團的水層。與RPLC相比,HILIC其他突出的優(yōu)勢包括“正交”選擇性(樣品制備兼容兩種模式),對質(zhì)譜具有更高的電噴霧離子化靈敏度(5-15倍),較低的操作壓力。
固定化多糖手性固定相
成功包覆的多糖手性固定相(CSPs)的改進版本在2000年代末實現(xiàn)。與早期的CSPs具有相似的多功能,但是對于腐蝕性的溶劑具有更好的穩(wěn)定性劑,可用于正相,極性有機和反相模式。
用于生物分子的色譜柱
80年代發(fā)展起來的大孔徑硅和聚合物填料可有效解決大型生物分子的分離。隨著重組蛋白,如單克隆抗體(mAb)等生物制藥的出現(xiàn),質(zhì)量控制中利用HPLC和毛細管電泳進行詳細表征的需求變得更加緊迫。最近,亞2μm微粒和核殼大孔徑顆粒以及一些創(chuàng)新的離子交換和尺寸排阻等材料也被證明可以有效的分離這些大分子的生物制劑。
高分辨率質(zhì)譜(HRMS),電霧式檢測器(CAD)和自動方法開發(fā)系統(tǒng)(AMDS)
在過去十年中,其他具有高影響力的進展主要體現(xiàn)在HPLC檢測和自動化領(lǐng)域。
高分辨率質(zhì)譜(HRMS)
HPLC與質(zhì)譜的聯(lián)用(LC / MS),集合了HPLC的分離能力和質(zhì)譜卓越的靈敏度和選擇性,已被視為完美的分析工具。LC/MS是雜質(zhì)和降解鑒定,藥物研究中的高通量篩選技術(shù)(HTS),生物分析試驗(生物流體中藥物和代謝物的LC/MS/MS檢測),藥物合成工藝放大在線監(jiān)測的首選技術(shù)。LC/MS已經(jīng)成為高效藥物清潔驗證以及潛在的基因毒性雜質(zhì)測定的標準技術(shù)平臺。過去十年中,HRMS(如TOF、OrbiTrap MS)和雜化質(zhì)譜(如Quadrupole-TOF或ion trap-OrbiTrap)得到了快速發(fā)展。HRMS和UHPLC以及二維LC的聯(lián)用使得代謝組學(xué)、蛋白質(zhì)組學(xué), De Novo蛋白測序和生物制藥表征等領(lǐng)域的研究愈發(fā)活躍。新興的個性化醫(yī)療領(lǐng)域中生物標志物測定和疾病診斷的通用技術(shù)平臺也許是LC/MS最令人興奮的機會。
電霧式檢測器(CAD)
缺乏理想的通用檢測器常常被視為HPLC的局限,盡管UV/Vis檢測器可以檢測具有發(fā)色基團的化合物。示差折光檢測器不適合梯度洗脫,敏感性不夠。蒸發(fā)光散射檢測器(ELSD)使用噴霧器技術(shù)與激光光散射檢測,是HPLC的一個選擇,也可以兼容梯度洗脫,但最近已經(jīng)被CAD(使用噴霧器和電暈放電檢測技術(shù))超越,CAD可以提供更好的靈敏度(低至ng)和更好的線性。CAD正逐漸成為藥物化學(xué)中的HTS、反應(yīng)過程監(jiān)控以及原材料/輔料測試的主流的檢測器。
自動化HPLC方法體系(AMDS)
復(fù)雜混合物的HPLC方法開發(fā)是一個很耗時的工作,因為需要優(yōu)化很多操作參數(shù)(柱尺寸,鍵合固定相和流動相A和B(有機溶劑/緩沖類型、pH值和離子強度)的類型,梯度洗脫時間和梯度范圍,柱溫度、流量)。一個常見的例子就是藥物活性成分(API)穩(wěn)定性指示分析或純度測定,其中所有的雜質(zhì)和降解產(chǎn)物必須分離,通過UV檢測器進行準確的定量。多年以來,基于模擬、預(yù)測、單純形優(yōu)化、柱/流動相篩選的軟件或自動化系統(tǒng)已經(jīng)促進HPLC方法的開發(fā)。雖然他們似乎還沒有很普及,但持續(xù)的改進提高了HPLC的性能和易用性。最新進入市場的是一個附加軟件包,兼容兩個常用的色譜數(shù)據(jù)系統(tǒng)。對于HPLC方法開發(fā)過程(優(yōu)化)中最耗時的部分,該軟件利用用戶定義空間的自動化序列方法來解決,其中使用了實驗設(shè)計(DoE)和質(zhì)量源于設(shè)計(QbD)的原則。導(dǎo)入完整的序列結(jié)果之后,該軟件還可以執(zhí)行統(tǒng)計分析,并顯示最佳條件。圖5說明了應(yīng)用在我們實驗室的該軟件的概念和特征。自DoE 和 QbD概念被接受以來,最初用戶表現(xiàn)出很高的興趣。AMDS對致力于早期藥物開發(fā)方法的實驗室特別有用。例如,為了支持利用多手性中心的復(fù)雜藥物分子進行API合成和藥物產(chǎn)品制造,10-20種具有代表性的HPLC方法(原材料、起始物料、中間體、最終的原料藥和藥物產(chǎn)品的非手性和手性方法,)接二連三地被開發(fā)出來。
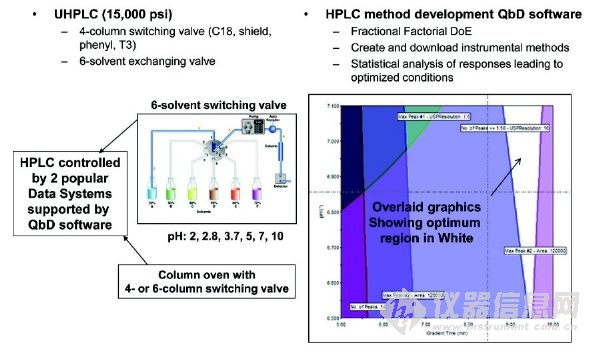
圖5 一個全自動方法開發(fā)系統(tǒng)的儀器構(gòu)造圖解,該系統(tǒng)使用QbD軟件和所得結(jié)果的圖形,顯示最佳分離(白色)的操作范圍設(shè)計空間。
結(jié)論
總之, 最近幾年中,HPLC仍然是一個高度有活力的領(lǐng)域,在儀器、色譜柱技術(shù)、應(yīng)用等方面有很多創(chuàng)新。醫(yī)藥科學(xué)家們最初將這些新技術(shù)應(yīng)用于研究、開發(fā)和質(zhì)量控制,他們是這項技術(shù)的早期采納者,同時也是受益者。UHPLC在研究與開發(fā)領(lǐng)域被快速的接受,并逐漸成為標準的UHPLC平臺,雖然在QC實驗室的應(yīng)用速度慢一些。新的色譜柱技術(shù)可以更快和更有效的分析復(fù)雜樣品、手性分子和生物分子。最后,UHPLC和2-D LC與高分辨率質(zhì)譜聯(lián)用技術(shù)的快速進展,已經(jīng)徹底改變了生命科學(xué)的研究,并將會在臨床診斷方面產(chǎn)生更大的影響。這些進展得到了在快速發(fā)展的藥物開發(fā)領(lǐng)域工作的分析化學(xué)家的歡迎。
對于LC在接下來的幾年的發(fā)展前景,一些創(chuàng)新的工作已經(jīng)被描述。就像Jim Jorgenson描述的:“更高的壓力(50000 psi)將使更小的顆粒和/或更長的色譜柱的使用成為可能,同時分離速度更快,效果也更好。由于熱量的產(chǎn)生和耗散問題,幾乎肯定的是需要使用sub-mm孔(毛細管)色譜柱。這并不是一件容易的事情,但是高速、高分辨率分離方面潛在的可能性是誘人的。”另外,在毛細管中填充極其均一的次微米二氧化硅粒子可以用來產(chǎn)生遠低于1μm的塔板高度,并能對蛋白質(zhì)變異體進行令人印象深刻的分離。這兩個研究結(jié)果還沒有準備好馬上成為主流分析工作或QC,但是第一個結(jié)果是鼓舞人心的,因此在這里值得一提。
原文作者:Michael W. Dong, Ph.D.
Davy Guillarme, Ph.D.